Historical overview
In 1901, Dr. Aloysius "Alois" Alzheimer and his colleague Dr. Emil Kraepelin examined a strange patient at the Städtische Anstalt für Irre und Epileptische, a long-established hospital in Frankfurt. The 51-year-old female patient, Auguste Deter, had gradually lost her short-term memory over a few years, and at the time of the study was unable to remember facts and figures even in the time span of a few minutes. She also showed significant personality and behavioural changes, the most striking of which were paranoid delusions. The patient attracted the attention of the then 37-year-old young doctor in an extraordinary way and he followed her up in the following years until her death on 8 April 1906. He and his colleagues then carried out a histological examination of the brain and, using Bielschowsky staining, described two curious pathological phenomena, plaques outside cells of amyloid origin and neurofibrillary tangles present within cells. In the autumn of 1906, he presented his results at the West German Psychiatric Congress in Tübingen and published them (Alzheimer 1907). In 1910, Kraepelin named the previously described disease Alzheimer's disease (AD) in the 8th edition of the Handbook of Psychiatry, which he edited, in the chapter on Presenile and Senile Dementia, and it became a well-known disease in medicine. In the 1990s, many people questioned whether Alzheimer had indeed described the disease known today as AK, until 1998, when it became possible to study the original microscopic specimens, confirming that the original identification was indeed correct. Until the 1970s, little was known about how the known neuropathological abnormalities could explain the symptoms seen until Katzman formulated the cholinergic hypothesis that the death of basal forebrain neurons and the reduction in the amount of acetylcholine they produce could underlie cognitive decline. This was followed in the 1980s by a more thorough understanding of the properties of the responsible proteins, the sequencing of beta-amyloid and the identification of tau in neurofibrillary tangles. In the 1990s, a major breakthrough was made with the discovery of the most important genetic risk factors, the amyloid precursor protein (APP), apolipoprotein e4 (apoE4), and presenilin 1,2 (PSEN1-2) genes. It was also during this period that society's attention turned towards the disease, as indicated by the first International Alzheimer's Day on 21 September 1994. The late 1990's also had therapeutic successes, with the introduction of the first interventional drugs, acetylcholinesterase inhibitors. The 2000's were marked by the introduction of imaging and biomarker identification techniques, which allowed the identification of key structural variations and the development of liqour diagnostic methods. Although there are still about 80 therapeutic trials in progress, the treatment options for the disease are extremely limited and research - on both pathomechanism and therapy - is expanding with more and more new chapters.

Alois Alzheimer

Auguste Deter
Epidemiology
The modern view of AD is that it belongs to the group of major neurocognitive diseases, a category that is a melting pot of some 60-100 neurological diseases of different origins. Among these, up to 20% are reversible forms, such as cognitive decline associated with psychiatric disorders (e.g. depression, generalised anxiety), vitamin deficiencies (e.g. vitamin B12 deficiency), metabolic abnormalities (e.g. Hypothyroidism), traumatic brain injury (e.g. subdural haematoma), toxic damage (e.g. hepatic encephalopathy) and cerebrospinal fluid circulatory disorders (e.g. normal pressure hydrocephalus). Recognition of these is essential as timely treatment can improve cognitive complaints.
However, progressive, irreversible forms, such as frontotemporal dementia, diffuse Lewy body dementia, Parkinson's dementia, corticobasal degeneration, etc., make up the bulk of the disease group. Definition of dementia, created by the National Institute on Aging in 1995, is still valid today: 'Progressive intellectual decline severe enough to cause impairment in daily routine and social activities'.
Among this group of diseases, AD stands out due to its prevalence, as it is estimated to be responsible for about 55% of cognitive decline in old age. In this context, it is not surprising that in the developed world, AD is the 6th most common cause of death, but it is by far the leading cause of long-term health loss and poor quality of life, mainly due to its progressive and chronic nature. Another significant problem is that while mortality associated with classical common diseases is decreasing, mortality associated with AD is increasing almost exponentially. The condition also places an enormous burden on the family members involved in the care of the patients. It is important to take into account the social phenomenon that, due to the prolongation of the childbearing years, most of these carers are members of the so-called sandwich generation, i.e. while caring for their elderly parents, they are also raising their own school-age children. According to US statistics, this affects around 16 million families, leading to more than 18 billion hours of unpaid leave each year (Alzheimer's Association 2016). All these factors combine to make it unsurprising that, in terms of health and social expenditure, AD is one of the world's 'most expensive' diseases, and also places a huge financial burden on society. For example, taking the above-mentioned country, the estimated total expenditure in 2016 was US$ 260 billion (Alzheimer's Association 2016). The huge prevalence and the significant social burden have made AD one of the greatest medical and social challenges of our time.
Causes and risk factors
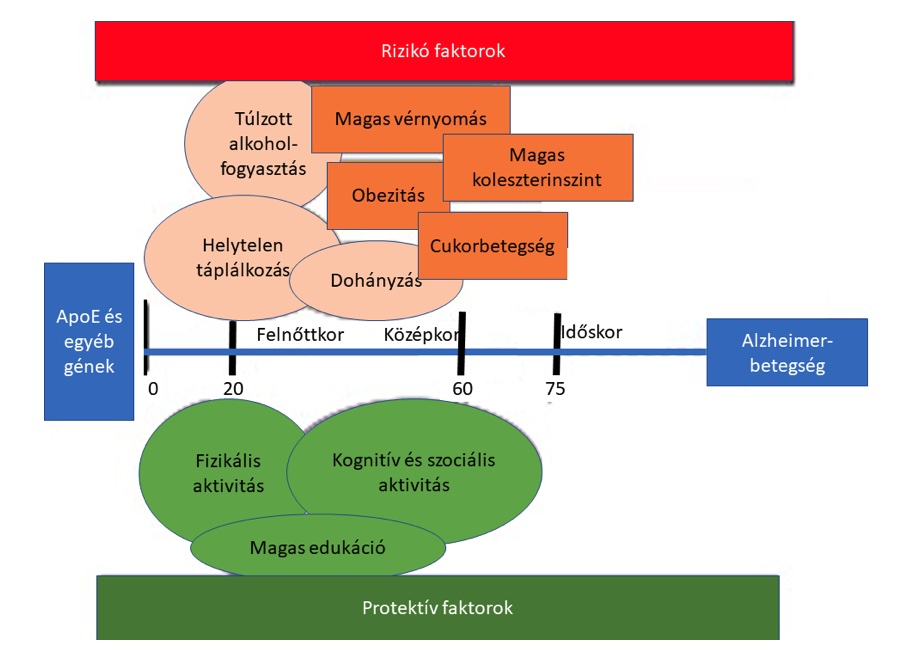
Unfortunately, despite intensive research, we still do not understand exactly what causes the development of the disease. In order to understand these, it is useful to distinguish between early onset Alzheimer's disease (EOAD) and late onset Alzheimer's disease (LOAD). Although the two forms of the disease are symptomatically and clinically similar, the underlying causes are significantly different. In EOAD, the underlying genetic mutation is either a hereditary familial accumulation - also known as familial AK - or a sporadic mutation. It occurs in about 1-5% of all cases of AK and is most commonly caused by mutations in the PSEN1, and less frequently in the PSEN-2 and APP genes. In LOAD, the problem is more complex, with a combination of genetic, environmental and lifestyle factors, similar to the Knudson double-hit hypothesis of cancer onset. Among the genetic factors, the best known is the role of the apoE4 allele, which is associated with a higher incidence and earlier onset of the disease. In addition, 33 additional chromosomal regions of potential risk were identified by whole genome analysis (GWAS). The importance of genetics is illustrated by the fact that Down's syndrome also has a much higher risk of developing AK.
Ageing itself is one of the most important factors, as the risk of developing the disease doubles every 5 years after the age of 65. This cut-off is also used to distinguish EOAD vs LOAD, the accuracy of which is disputed by many. Other significant risk factors are classic cardiovascular factors, hypertension, type 2 diabetes, smoking, obesity, high blood cholesterol, high blood fatty acid levels, high blood homocysteine levels (Lindsay et al. 2002). The role of traumatic brain injury, which is mainly studied in athletes, is the subject of interesting and intensive research. Head trauma also appears to lead to the development of AK or Parkinson-like dementia, collectively referred to as chronic traumatic encephalopathy. The description of the phenomenon itself is not new, published in 1928 by Dr. Harrison Stanford Martland in boxers, named as dementia pugilistica. Other types of neurological involvement may also be risk factors, of which affective disorders are prominent. The dementia-like appearance of psychiatric illnesses is also reflected in their classical nomenclature, such as dementia praecox in schizophrenia and pseudodementia in major depression. It is also well known from psychiatric practice that severe forms of anxiety and mood depression are associated with a significant decline in cognitive function. It should be noted that the chronic use of certain medications, notably benzodiazepines, anticholinergics and proton pump inhibitors, also increases the risk. Lifestyle factors are also very important, as higher education, knowledge of foreign languages, multiple mother tongues, sustained mental and physical activity and a balanced diet have been shown to be protective factors. The importance of these factors is illustrated by the large number of studies addressing this problem (e.g. the FINGER study) and estimates that millions of cases of AK could be prevented each year by precise control of these factors.
In conclusion, the development of the disease requires a combination of several factors that alter the metabolic processes in the brain: reduced blood supply to the neural tissue, cerebral microbleeds, pericyte migration, inflammation, mainly with activation of microglial processes and a characteristic rearrangement of the cytokine profile towards inflammation, altered lipid transporters and impaired amyloid clearance mechanisms. At the end of the different processes, however, a very similar neuropathological picture emerges: amyloid plaques are deposited between neurons and phosphorylated tau bundles in neurons.
However, the appearance of the two proteins is different in time and space. The amyloid appears earlier in the disease, primarily in the mesolimbic areas, hippocampus and basal forebrain, and then gradually affects the entire cortex. The tau is thought to appear later in the disease, typically in the transentorhinal cortical areas, then in the limbic areas and finally in the neocortex. The pattern of onset and spread has been described by Braak's group.
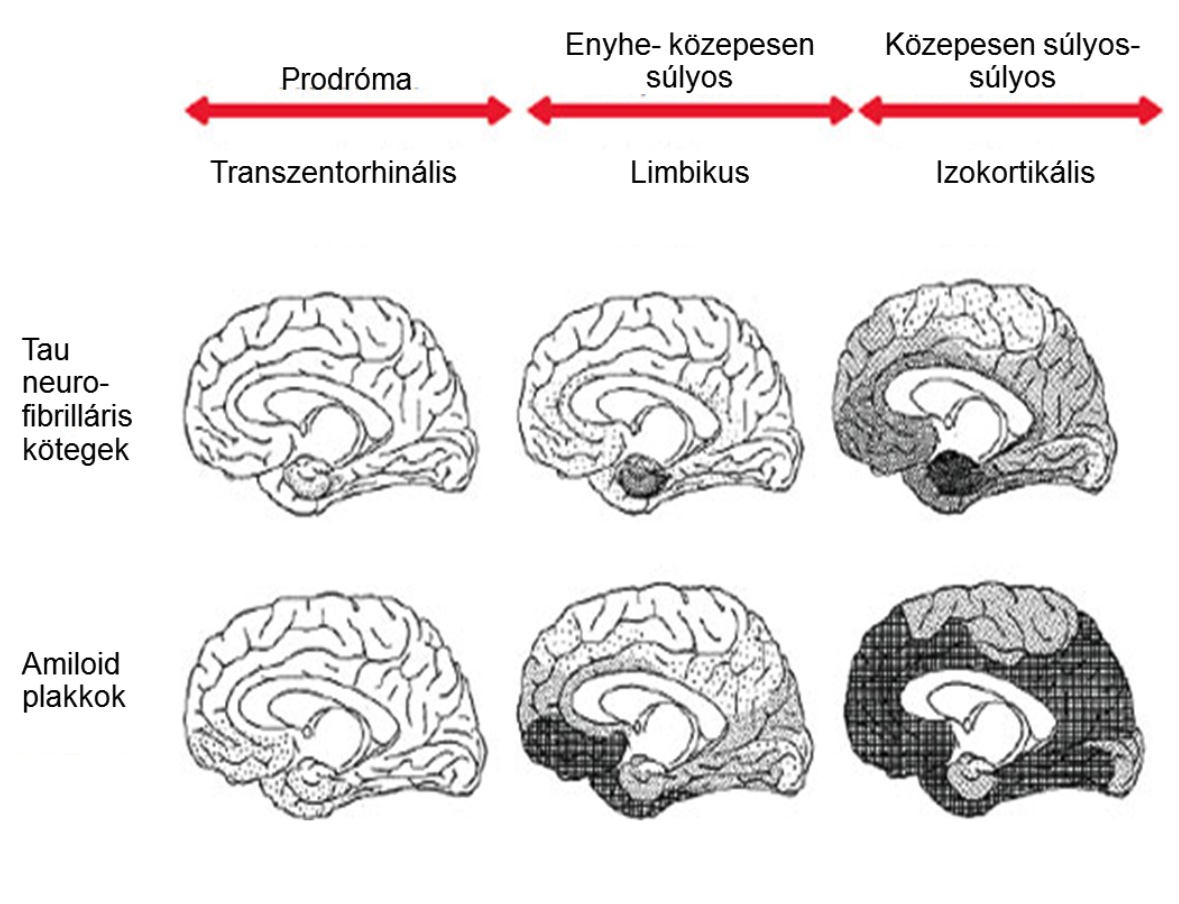
Stages of tau and amyloid deposit propagation described and illustrated by Braak (Braak and Braak 1991).
During propagation, neurons die due to deposition of proteins with altered structures, and the protein that is discharged from the cells is taken up by new neurons, thus passing the abnormal pattern on to each other. It is important to note that it is a relatively recent discovery that the appearance of proteins, especially amyloid, precedes the first symptoms by decades, so that certain biomarkers may already show positivity at this time.
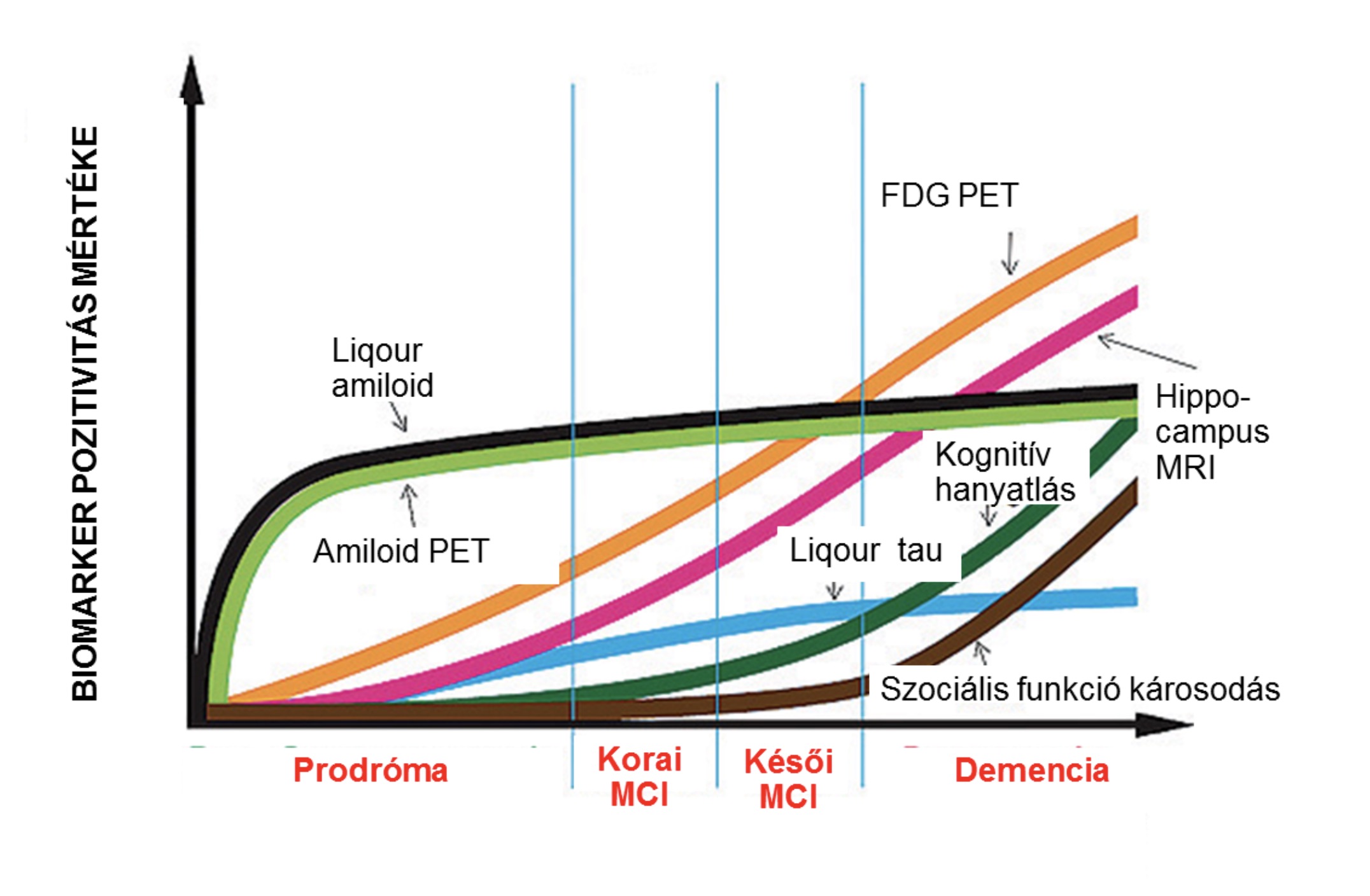
Changes in the predictive value of key biomarkers of Alzheimer's disease during disease progression (Source: Alzheimer's Disease Neuroimaging Initiative- http://www.adni-info.org/Scientists/ADNIOverview.html).
Interestingly, the appearance of abnormal proteins and the mode of spread is strikingly similar to the picture seen in prion diseases, which led to the AD prion hypothesis. Currently, several studies demonstrate that the injection of pathological proteins into the nervous system can lead to the development of the disease. The question of the infectious origin and the role of the intestinal flora in the development of the disease remains an open question in science and is subject to intense debate.
Symptoms
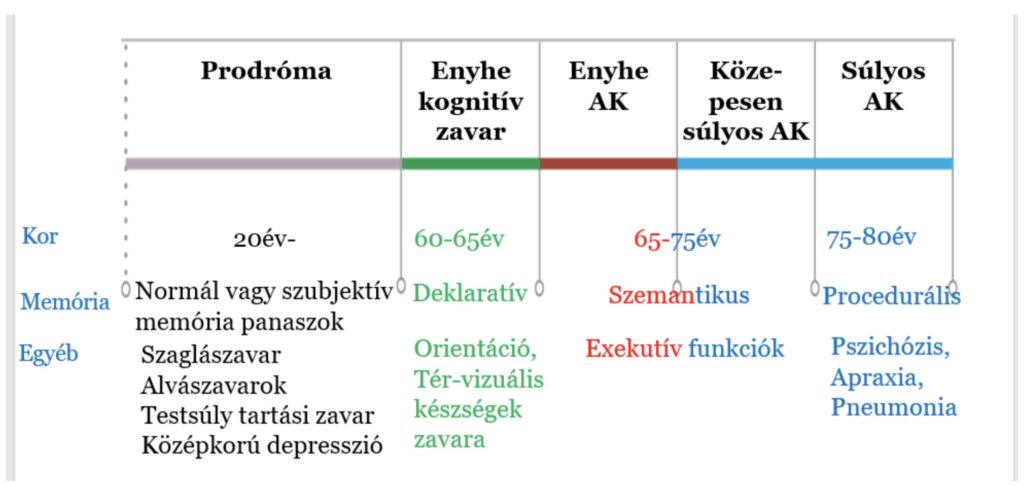
The first symptoms typically begin around the age of 65 years, an earlier onset is relatively rare and typically occurring in variants of genetic origin. The course of the disease is approximately 4-8 years from the onset of the first symptoms, but there is considerable variation, up to 20-22 years. However, an important phenomenon is that the prodromal phase, which is the focus of intensive research, starts decades before the clinically evident symptoms appear, in line with the pathological phenotypes. However, it appears that, although memory complaints are not detectable at this stage, impairment of autonomic and limbic functions may already be evident. Of these, an impaired sense of smell (hyposmia), the appearance of sleep disturbances, loss of weight control (the weight paradox in AD means that although being overweight is a risk factor, patients lose weight before the onset of the disease, so it may falsely appear that excess weight is a protective factor for AD as they age) and depression in middle age. The opinion about depression as a prodrome or risk factor for the disease, is not uniform, but a consensus is emerging that early onset is more of a risk factor, while early onset in old age is more of an early marker of the onset of the disease.
The prodromal phase is followed by a period of mild cognitive impairment, referred to in the literature as MCI. MCI means that the affected person experiences a decline in cognitive function but is able to compensate, so that there is no cognitive deficit and the definition of dementia is not met. It is estimated to affect around 20% of the population over 65 years of age. It is important to note that although the presence of MCI is associated with a higher risk of developing dementia, an actual cognitive deficit does not always develop, and the MCI status may stagnate or even improve. Detecting this stage is essential as it can help in early patient identification and timely introduction of therapeutic interventions. The clinical diagnosis of MCI is based on the Petersen criteria.
During the progression of Alzheimer's disease, cognitive and neuropsychiatric symptoms follow a characteristic sequence. In the early phase, short-term memory impairment is the most common complaint. Characteristically, declarative episodic memory (factual, consciously recalled e.g. shopping list) content associated with the hippocampus is most affected, while semantic (e.g. experiences) and procedural memory (e.g. execution of movement patterns) associated with neocortical areas is preserved. Remarkably, long-term memory is spared. Characteristically, therefore the ability to memorize (imprint) new memory content is lost. This is often accompanied by an orientation disorder, which usually affects spatial orientation more (e.g. getting lost on the way home from the shop) than temporal (e.g. what month is it). It may be characteristic that, when talking to the patient, a previous transient episode of amnesia may be recalled (the patient seems to have lost a period of the day).
In the moderate stage, semantic memory (e.g. recognising family members) is also affected, and typically patients lose executive functions (e.g. paying, managing, taking medication). As part of the frontal lobe symptomatology, the ability to acknowledge their disease is also impaired. Behavioural changes are also common: apathy (severe depression), agitation (irritability), aggressive behaviour, even hallucinations and psychosis. In this way, patients progressively lose the ability to take care of themselfs.
In the severe stage, all cognitive functions are affected and motor functions are usually impaired, this impairment can be present even in the form of severe myoclonus. The patient loses the capacity for self-care and orientation and becomes bedridden. In severe AD, the most common cause of death is pneumonia.
Diagnosis
The diagnostic protocol for the disease has changed several times over the past decades, unfortunately making it difficult to compare the results of current clinical trials with older analyses. It is important to note that, although the diagnostic repertoire has expanded, a definitive diagnosis can still only be made by histological examination. Only a probable diagnosis of AD can be made on the basis of clinical signs and data. The diagnosis requires first and foremost proof of dementia, for which it is almost sufficient to know the concept described earlier: cognitive decline affecting social life and showing progression. Of course, other psychiatric and physical causes must be excluded, for which extended laboratory tests and imaging studies are essential. A neuropsychological test is recommended, which should cover at least 5 cognitive functions and at least 2 should be affected: memory, complex executive function (e.g. writing, sentence production), spatial and visual skills, language function, behaviour and personality changes. Completing a neuropsychological test also helps to identify various neurocognitive disorders. The most commonly recommended test packages are the Addenbrooke's Cognitive Examination (ACE), Montreal Cognitive Assessment (MoCA), Alzheimer's Disease Assessment Scale-Cog (ADAS-Cog) and the Mini-Mental Test (MMS), with recommended cut-offs. Of these, the MMS is less sensitive and is now recommended for primary screening and staging, but is nevertheless one of the most widely used dementia screening tests, with a score below 25 being a good indicator of dementia. For staging, the Clinical Dementia Rating Scale (CDR) is also an excellent tool.
Once the presence of dementia has been established, the likelihood of AD requires a prolonged onset, confirmed progression (based on anamnesis, possibly supported by repeated neuropsychological tests), typical symptom onset (amnestic short-term episodic memory impairment, later on, difficulty in word learning or spatial, visual impairment or executive dysfunction) and the exclusion of other causes, for which structural MR is an essential requirement. In addition to general brain atrophy, this should be analysed for bilateral temporal lobe-cerebellar predominance cortical atrophy and hippocampal volumetric atrophy. The diagnosis may be confirmed by certain cerebrospinal fluid biomarkers, such as low Ab-42 or Ab-42/Ab-40 ratios, or elevated tau and phospho-tau (p-tau) values. If available, positron emission tomography (PET) biomarkers (amyloid, tau) can clarify the diagnosis. The criteria described are the NIA-AA (National Institute on Aging and Alzheimer's Association) criteria for Alzheimer's disease.
The other diagnostic system currently in use is the Diagnostic and Statistical Manual of Mental Disorders V (DSM-5), developed by the American Psychiatric Association (APA), which favours the use of the term major neurocognitive disorder rather than dementia to blunt the stigmatising concept of dementia. The priority given to memory impairment has been reduced in the diagnosis, recognising that in many cases, AD begins in a so-called non-amnestic form (e.g. speech impairment, executive dysfunction). In cases where social independence is preserved but deficit-level impairment in cognitive function is detected, mild cognitive impairment (MCI) is considered. If social activity is already affected and the person is dependent on others for daily activities, a diagnosis of definitive neurocognitive disease can be made (essentially the classic definition of dementia). If biomarkers show incidental positivity and the person does not produce symptoms, the diagnosis is prodromal AD. The DSM system therefore focuses on symptom progression, which may be important for early biomarker identification studies and intervention trials.
Basic criteria
Supportive criteria
A)
Early and significant episodic memory impairment with the following features:
Significant, progressive change in memory function over a period of at least 6 months as observed by the patient or a relative
Episodic memory impairment objectively identified by neuropsychological or clinical examination
The episodic memory impairment was the first symptom of PD, either alone or accompanied by other complaints
Supportive criteria
B)
Medial temporal lobe atrophy
Bifrontal-bitemporal atrophy with volume loss of hippocampus, amygdala, entorhinal cortex by visual analysis of skull MRI or quantitative volumetric measurement.
C)
Abnormal liqour biomarker values
Decreased amyloid beta 1-42 concentrations, elevated tau or p-tau values. Any other validated CSF biomarker
D)
PET biomarker positivity
Decreased glucose metabolism bilaterally in temporo-parietal areas. Other validated PET tracer (amyloid, tau PET)
E)
Evidence of autosomal dominant AD mutation
Therapy
According to our current knowledge, AD is not curable. However, it is extremely important to stress that this is not the same as being untreatable, thus providing a better quality of life for the patient and the affected relatives. However, proper diagnosis and identification of the stage of the disease are essential to ensure optimal treatment options. Failure to recognise the disease may hinder timely therapy or make it impossible to participate in ongoing drug research. Failure to perform the necessary tests for a differential diagnosis may lead to overlooking an underlying treatable cause (e.g. failure to perform thyroid function tests) or to resort to a non-recommended therapy in the presence of another degenerative disease, to the detriment of the patient (e.g. use of a neuroleptic in diffuse Lewy body dementia). In this light, it is always necessary to consider whether the recommended diagnostic algorithm has been followed before introducing or changing therapy.
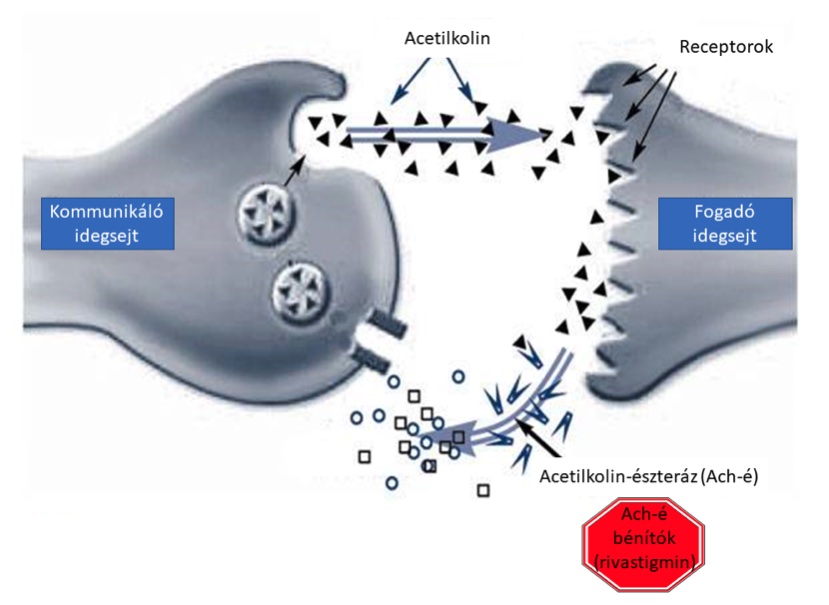
In addition to the use of appropriately chosen medications, therapy should also include psycho- and socio-therapeutic considerations. As the patient's willingness to cooperate is reduced, thorough information of the relative and discussion of treatment options are of utmost importance. The most commonly used drugs are cholinesterase inhibitors such as donepezil, galantamine and rivastigmine. These are typically recommended in mild to moderate stages. From moderate stage onwards, therapy can be supplemented with memantine, an N-methyl-D-aspartate (NMDA) receptor inhibitor. Another important factor is the appropriate therapy of the associated psychiatric disorders, which are often particularly difficult to manage, especially sleep disturbances and agitation. In addition to drug therapy, rehabilitation interventions such as diet optimisation, regular exercise, speech therapy, motor and cognitive training are increasingly important. Non-pharmacological interventions are also particularly important because in the last 2 decades more than 100 phase III trials have had to be discontinued due to failure. This is one of the reasons why the focus on curing the disease has recently expanded to include other co-morbidities in AD.
New drug development trends have also emerged in recent years. One of the therapeutic options to reduce the severity of Alzheimer's disease is the prevention of β-amyloid plaque formation. Three molecules are currently being investigated for their anti-aggregation effects: tramiprostate, colostrinin, sciloinositol. Tramiprostate inhibits the binding between amyloid and glucosaminoglycans, thus keeping the amyloid protein in monomeric form. The ability of colostrinin to inhibit amyloid aggregation has so far only been demonstrated in animal studies, but the results so far are promising. The efficacy of sciloinositol has also only been demonstrated in animal studies in mice. It is able to stabilise the already aggregated amyloid, thus preventing its toxic effect on neurons.
Another possibility is the development of agents that inhibit changes in tau protein structure. Phenothiazine, methylthioninium chloride (methylene blue) have been shown to inhibit tau aggregation in cell culture experiments, but have also been used in the treatment of Alzheimer's disease, although significant results have only been achieved using methylene blue. In a series of treatments over six months, cognitive function in Alzheimer's patients improved steadily. Among the agents that inhibit the phosphorylation of tau protein, kinase inhibitors are considered to be of major importance. Glycogen synthase kinase-3, of which lithium is the most studied inhibitor, appears to be a potential candidate. In addition, other agents could be considered, including pyrazolopyridine and agents containing the active substance valproic acid.
Immunotherapy is a therapeutic strategy being investigated by most pharmaceutical companies. Two types of immunotherapy are known: active (vaccination) and passive (use of monoclonal antibodies). Active immunotherapy is more concerned with processes that inhibit β-amyloid aggregation, whereas passive immunotherapy seeks to prevent changes in the structure of the tau protein. So far, passive immunisation seems to be the more effective method in terms of research results, but neither therapeutic option can be neglected in the future.
Prevention
The Finnish Geriatric Intervention Study (FINGER-study) was conducted between 2012 and 2014 to analyse early intervention therapy options for the prevention of dementia. The 4-pillar study involved 1260 participants aged 60 to 77 years and included dietary changes, physical activity, cognitive and social training, and control of cardiovascular and metabolic risk factors. The diet was based on the official Finnish nutritional recommendations, and physical activity therapy was guided by physiotherapists and included the development of muscle strength and endurance, and the improvement of balance. The WHO recommended physical activity of 150 minutes/week was used. Cognitive training consisted of 10 sessions of 60-90 minutes of training led by psychologists. This was followed by an individualised computer-based test package, specific to the weaker cognitive domains, which participants used 3 times a week for 10-15 minutes. Risk factors were controlled in the light of current health recommendations. At the end of the study, a significant difference was identified between the control and intervention groups, with those included being significantly less likely to develop dementia.
Modern intervention trials highlight the desirability of identifying the disease and its risk factors from the earliest possible stage, as an early and appropriate prevention strategy can significantly reduce the chances of developing a serious condition. Of course, even in the symptomatic phase, it is important to complement drug therapy with lifestyle interventions. Presumably, new drug and preventive options will emerge in the future. A prerequisite for this is a thorough understanding of the pathological and comorbid factors of Alzheimer's disease.
The role of sleep
Over the past decades, our understanding of the function of sleep has changed dramatically: whereas it was once thought of as a passive process to replenish resources, it is now increasingly seen as an active mechanism that is a cornerstone of brain adaptability. However, the structural integrity of sleep is a fundamental requirement for its function in learning and memory processes. The most common way to study sleep structure is by polysomnography (PSG), in which brain and muscle activity, eye movements, respiratory activity and heart rate are recorded simultaneously. Using brain activity analysis, sleep can be divided into different stages: rapid eye movement (REM) sleep and non-REM (NREM) sleep, which consists of three stages (S1, S2, S3). Each stage has its own distinctive identifying signs, such as K-complexes and sleep spindles, which are characteristic of the S2 stage. The S3 stage is also known as slow-wave sleep because of its characteristic wave activity.
Sleep changes are a well-known feature of the normal ageing process, during which sleep is fragmented, sleep duration is prolonged, the number and length of night wakings increase, and daytime sleepiness is increased. Sleep disturbances due to sleep-wake cycle disruption, sleep-dependent breathing and leg movement disorders such as obstructive sleep apnoea syndrome, restless leg syndrome, periodic leg movement disorder syndrome become common. The amount of slow-wave sleep decreases, while the length of S1 and S2 phases lengthens in response. The REM phase is characteristically preserved. Another characteristic change is a reduction in the number of sleep spindles and K-complexes.
An important observation is that sleep disturbances may also be a risk factor for AD. In insomnia, it has been described that amyloid plaques characteristic of AD pathology appear in parallel with sleep deprivation. Furthermore, sleep problems are undoubtedly a sub-symptom of AD, with increased abnormalities characteristic of the ageing process, and REM phase involvement has also been observed. Presumably, the involvement of this phase may also indicate the progression of the disease, as it is normal in the mild stage. In conclusion, the study of sleep is a promising area of AD research that may help to understand the underlying causes of cognitive impairment.
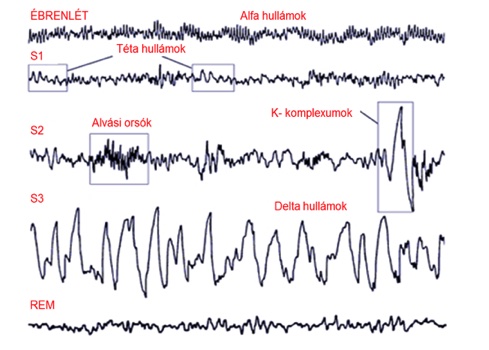
EEG patterns and sleep stages characteristic of human sleep structure.
The role of epilepsy
Epilepsy is the 6th most common neurological disorder, characterised by intermittent recurrent epileptic seizures. Its incidence peaks in childhood and in old age. During epileptic seizures, nerve cells are activated in an abnormally strong coordination, resulting in the simultaneous electrical discharge of large groups of cells. The clinical features of the seizure are determined by the area of the brain where the affected nerve cell group is located and the direction in which the abnormal activity can spread from there. Seizures are accompanied by convulsions (stiffening followed by convulsive shaking) in 60% of cases, but in the remaining 40% they are not accompanied by any obvious movement symptoms. The abnormal activity may affect the whole surface of the cerebral cortex, in which case it is called a generalised seizure, or it may be confined to certain areas of the cortex, called focal epileptic seizures. The symptoms during a seizure are determined by the function of the affected cortical area. After the seizures, symptoms resulting from temporary exhaustion of the cortical areas are typical, for example Todd's palsy in the case of seizures in the motor cortex, or confusion, psychosis as a symptom of the temporal or frontal lobe. However, the epilepsy disease is more than a collection of seizures. Epileptic dysfunction leads to structural and molecular changes in the affected brain tissue. These changes lead to increased epileptogenicity and the development of epileptic seizures. Between seizures, brain activity also deviates from normal and is characterised by abnormal discharges of typically short duration that affect the integrity of brain network functions. These are collectively referred to as interictal epileptiform phenomena. The quality of life of people with epilepsy is primarily determined by the frequency of seizures, so the main goal of medication is to achieve seizure freedom. However, new research suggests that interictal phenomena are also of particular importance, as they are most closely associated with the cognitive impairment often associated with epilepsy.
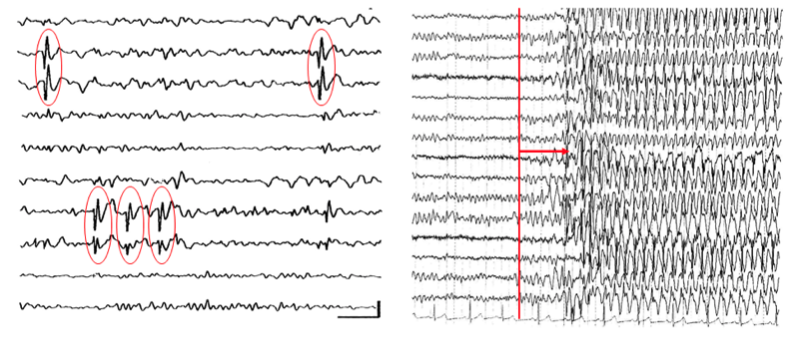
Interictal discharges on the left side of the figure, while generalised seizure EEG image on the right side of the figure.
The link between the two diseases is a relatively new area of research, only coming to the fore in the last decade, despite previous studies have found overlaps.In 1892, Paul Blocq and George Marinesco were the first to describe the presence of amyloid plaques in human brain tissue, using samples from surgical specimens of epileptic patients. Later, the presence of amyloid in brain tissue from epilepsy patients was confirmed and another protein, tau, was detected in these samples. In a pathology paper published in 2016, temporal lobe sections from 33 therapy-naïve epilepsy patients identified phosphorylated-tau bundles in 94% of the samples (Tai et al. 2016). Other identities were also described, e.g. the reduced hippocampal volume characteristic of AD was also detected in patients with temporal lobe epilepsy. Reduced functional connectivity and resting-state activity in default mode network (DMN) were found in both diseases by functional magnetic resonance imaging (fMRI) scans. PET scans depicted reduced basal temporal lobe area function in both cases. Considering that the primary brain area affected in AD is the hippocampus, the most epileptogenic area of our brain and the source of the most common type of epilepsy, temporal lobe epilepsy (TLE), the similarities are not surprising. Currently, a number of clinical studies in humans have also demonstrated that epileptic seizures are more frequent in Alzheimer's disease and that they impair disease survival and quality of life in patients. At the same time, a number of drug trials have been launched on the use of anti-epileptic drugs in the treatment of AD.
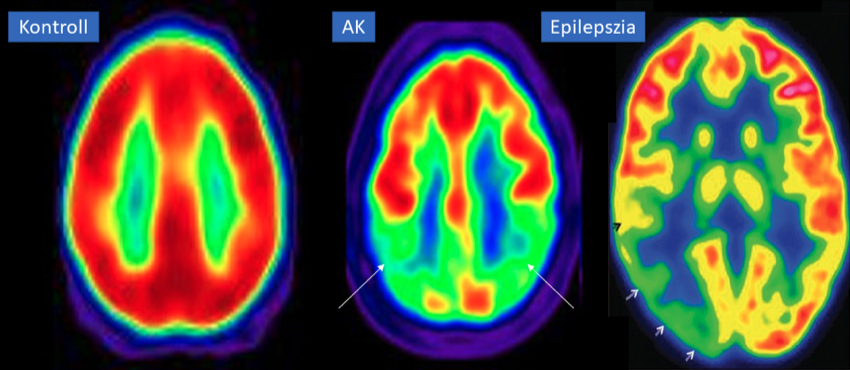
PET scan of a patient with Alzheimer's disease and epilepsy. Arrows indicate that both patients have a significant reduction in temporal lobe metabolism.
The role of inflammation
Inflammation is a biological response in which protective, sentinel mechanisms are activated in response to the presence of a potential cell damaging factor. The aim of a sudden, acute inflammatory response is to remove the abnormal element as quickly as possible and restore the body to a healthy state. If the factors that stimulate the inflammatory response persist or the processes that regulate it are disturbed, chronic inflammation develops. During the chronic response, tissues are damaged and cells can be formed that attack the body's own cells: an autoimmune process that is involved in the development of many diseases. The inflammatory hypothesis of Alzheimer's disease was formulated by Krstic and Knuesel in 2013, opening a new line of thought in the understanding of the disease, in addition to the previous theories (the amyloid cascade theory outlined by Hardy and Allsop, the Bartus cholinergic hypothesis, etc.).
A specific form of inflammatory response is neuroinflammation in the brain caused by tissue damage or infection, during which a special defence cell in the brain, the microglia, is activated. The microglial cell itself is the main immune cell in the brain, a type of cell belonging to the group of macrophages that cleans up dead cells and cellular debris in the brain. It circulates in the tissues in a resting state, and then, when threatened, it proliferates, becomes activated and tries to ward off potential danger by engulfing it - phagocytosis. At the same time, it stimulates the production of inflammatory molecules (cytokines), the most important of which are interleukin-1β and 6, tumour necrosis factor-α and transforming growth factor-β. Interestingly, during prolonged microglial activation, its phagocytosis activity is reduced, similar to that seen in other diseases; for example, in chronic airway inflammation, the activity of cells capable of phagocytosis is reduced, making patients more susceptible to infections. Since this function is essential in the breakdown of the abnormal proteins, amyloid plaques and tau tangles, which are so characteristic of Alzheimer's brain tissue, it is assumed that chronic inflammation is also a key factor in the deposition of these proteins, in addition to damaging neurons.
This is supported by research on another group of enzymes involved in the regulation of inflammation, matrix metalloproteases (MMPs). These enzymes break down damaged tissue and promote the regeneration and reformation of blood vessels and nerve tissue in its place. During chronic microglial dysfunction, their regulation is disturbed, resulting in increased damage to nerve tissue. Unfortunately, in the environment of abnormal amyloid and tau, the activity of MMPs is further enhanced, so that under chronic inflammation, pathological protein accumulation has multiple effects on neuronal cell death. This is further confirmed by the fact that microglia are also significantly activated in the environment of amyloid and tau, as they are recognised as pathological factors.
Chronically activated microglial cells thus maintain a permanent inflammatory response (MMP dysregulation and increased inflammatory cytokine production), but are no longer able to perform their original cleansing (phagocytosis) function, which may be an important factor in the disease. Another group of glial cells, astrocytes, also known as stellate cells because of their shape, are the most populous cell type in the central nervous system, accounting for 25 percent of the brain volume. Their functional role has long been underestimated, but we now know that their presence is essential for many important physiological processes. They are involved in strengthening neuronal connections, creating and maintaining the blood-brain barrier, and in the maintenance of ion balance and a normal environment for neurons, as well as being responsible for glutamate metabolism.
Interestingly, post-mortem histopathological studies have shown an increased presence of astrocytes in Alzheimer's disease deaths. The phenomenon, reactive astrogliosis, is well known and is an inherent part of all inflammatory processes involving nerve cell damage. Astrogliosis is manifested by an enlargement and proliferation of stellate cells and increased production of the acid fibrillary glial proteins (GFAP). The direct effect of this reactive process on the accumulation of these abnormal proteins is still controversial, but it is known that the α1-antichymotrypsin protein, regulated by activated astrocytes, affects beta-amyloid deposition and tau phosphorylation. For us, however, the correlation between the extent of astrogliosis and mental decline provides the most important evidence that astroglia play an important role in the disease and may be involved in the inflammatory response.
Recent research has shown that if microglial activity is sustained for a long period of time, it can lead to an increase in the function of a type of cell, the type A1 astrocyte. Such astrocytes, unlike others, do not nourish or protect nerve cells but stimulate their death. Nerve cell death can also occur indirectly. It has been observed that reactive astrocytes cannot fully perform their supportive function, making nerve cells more vulnerable to external influences, to the generation of free oxygen radicals and to the cell-damaging effects of excessive glutamate stimulation.
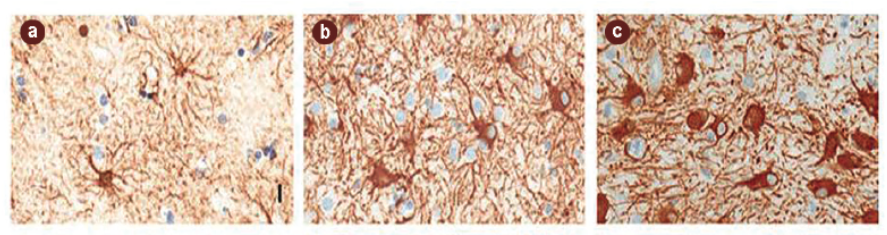
Accumulation of astrocytic glia (brown staining) following inflammation in the neuronal environment in post-mortem histological images of Alzheimer's disease patients.
Theory of infection
The fact that Alzheimer's disease develops late in life has raised the possibility that it may be caused by a previously acquired, slow-onset infection. The clinical similarities between syphilitic dementia and Alzheimer's disease were suggested by Alois Alzheimer himself, and then Perusini and Bonfiglio confirmed similar histological lesions by pathological studies. Subsequently, interest has focused on pathogens that are neurotropic, i.e. that primarily attack and persist in the nervous tissue and cause slow, prolonged infections that can last for decades. The primary hypotheses have been confirmed by modern pathogen isolation studies that have shown an increased presence of certain infectious microorganisms in the brain tissue of Alzheimer's disease patients. Viruses have been confirmed to be herpes viruses (e.g. cytomegalovirus, human herpes viruses 1 and 2), bacteria Chlamydia pneumoniae, Helicobacter pylori, Borelia burgdorferi responsible for Lyme disease, Treponema pallidum causing syphilis and certain oral bacteria. Among fungi, Candida albicans is the focus of interest.
The pathway of brain tissue disease is thought to be multifaceted. These pathogens have the ability to cross the blood-brain barrier to reach the central nervous system where they can trigger a chronic inflammatory response and promote the development of Alzheimer's disease. This is based on the fact that their entry is primarily mediated by microglia, a key factor in Alzheimer's disease. The body is usually able to destroy them, but their ability to persist for long periods of time is based on the development of strategies to easily hide or evade immune attacks. In the case of reduced defences for whatever reason (e.g. older age), the chances of this happening are greatly increased.
Another possible mechanism is that the pathogens actually generate an immune response in peripheral areas that mobilises the whole body, whereby immune cells present in the periphery flood the central nervous system and then act in a manner reminiscent of microglia. This is confirmed by studies that have shown an increased incidence of dementia in elderly people who have suffered from chronic recurrent infections.
However, it is important to clarify that, as far as we know, no single pathogen is responsible for Alzheimer's disease, and these mechanisms are thought to be only a partial manifestation of the cause. Nevertheless, the role of infections in the development of neurodegenerative diseases remains an important line of research.
One possible hypothesis for the disease-causing role of infections is the reactivation of latent herpes infections. This was first proposed in a 1982 study, which found that brain tissue affected in the first stage of Alzheimer's disease overlapped well with areas where human herpesvirus 1 (HS1) was present. The virus is carried by 80 per cent of the human population and after primary infection it hides in the brain nerve roots where it survives. Periodic reactivation occurs when the immune system deteriorates. This can be seen during periods of psychological and mental stress or illness, when the herpes virus reappears, causing cold sores. If there are predisposing factors to Alzheimer's disease (e.g. Apo-e4 genetic defect), the herpesvirus plays a role in the abnormal formation of the aforementioned amyloid and tau proteins by causing chronic microglial function by re-igniting them. The development of antiviral therapies or vaccines to inhibit viral replication is a promising direction, but these drugs have many side effects and reliable vaccines against herpes virus are unfortunately still limited.
Among the many bacteria, Spirochetes are of particular interest due to their ability to reach brain tissue via lymphatic vessels and nerve fibres, in addition to the bloodstream. The best known of these are the pathogens of syphilis and Lyme disease mentioned above. These are characterised by the fact that they can enter the brain via the olfactory nerves, even through the mucous membrane of the nasal cavity. Several studies have demonstrated the presence of these bacteria in the olfactory nerve cells of Alzheimer's patients in the early stages of the disease. This is particularly interesting in view of the fact that olfactory dysfunction in neurodegenerative diseases often precedes the onset of mental decline.
A further characteristic of Spirochetes is that chronic inflammation very often develops after acute infection, due to their plaque-forming property: they are able to build up a plaque around themselves that protects the pathogen from the immune system. Chronic inflammation favours the deposition of abnormal proteins through microglial function. Interestingly, Spirochetes have a protein that directly stimulates amyloid formation. Several species of this group of bacteria are normally present in our body and are common components of the oral flora.
Our oral flora is incredibly diverse, home to around 900 species of bacteria, according to molecular studies. Interestingly, there is considerable individual variation in the type and number of species, with up to 200 bacterial species. The importance of the microbes that live with us - the microbiome - is being demonstrated in an increasing number of diseases, Alzheimer's disease being no exception. The diversity of the oral flora is an important fact in the light of the high prevalence of the most common oral inflammations (dental caries, periodontitis, gingivitis), affecting 60-80% of the population, mainly due to changes in dietary habits and poor oral hygiene. These inflammatory processes, in particular periodontitis, significantly alter the composition of the oral flora, favouring the proliferation of bacterial species that can survive in the absence of oxygen. Among these, Treponema denticola, Tannerella forsythia and Porphyromonas gingivalis are important species in the development of Alzheimer's disease. Studies have shown that these microbes are able to enter brain tissue through the lymphatic vessels and the nerves that innervate the teeth. Subsequent studies have shown that patients who were found to be infected with these bacteria were more likely to develop Alzheimer's disease. It has been suggested that the bacteria's cell wall contains a substance (lipopolysaccharide) that induces the production of pro-inflammatory cytokines, which leads to persistent activation of microglia. The bacteria are able to survive within the brain tissue, acting as a chronic inflammatory focus. The importance of oral inflammation is increasingly being demonstrated in a growing number of studies. A large cohort (597 subjects), 32-year follow-up study in 2010 demonstrated a strong association between the onset of dementia and tooth loss, gingival bleeding, depth of pockets around the tooth and loss of the bony sockets around the tooth. Oral hygiene appears to be a highly important, modifiable risk factor for Alzheimer's disease.
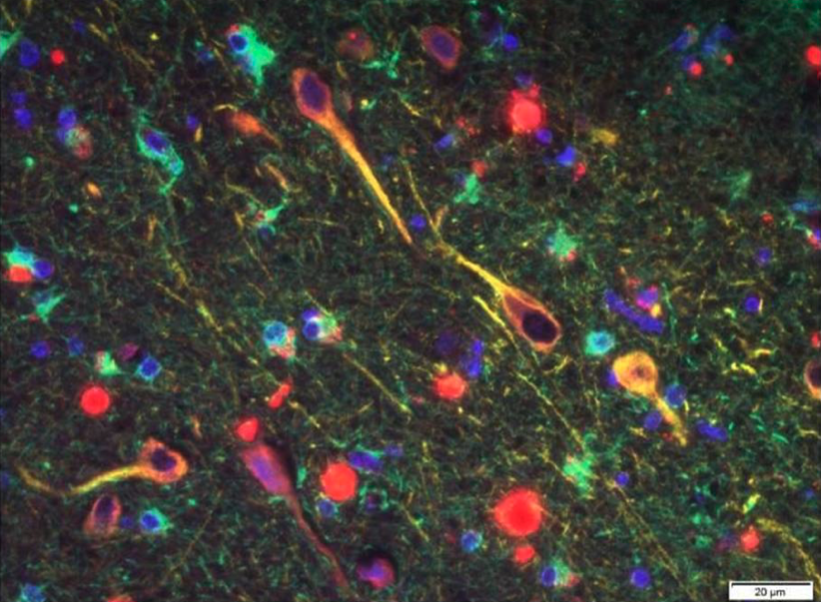
Histopathological detection of Porpyhriomonas gingivalis bacteria in brain tissue of Alzheimer's disease patients.